4th gen EGFR inhibitor
BI-4732
BI-4732 is a potent and novel fourth-generation epidermal growth factor (EGFR) inhibitor, suitable for in vitro and in vivo studies. It exhibits potent antitumor effects against on-target resistant EGFR mutations, including C797S, as well as EGFR-activating mutations (E19del and L858R). BI-4732 also displays efficient blood–brain barrier penetration capabilities and robust antitumor activity within the CNS. It is closely related to BI-8128, also available on opnMe.



More information
EGFR is a transmembrane tyrosine kinase receptor that plays a pivotal role in cellular processes such as proliferation, differentiation, and survival. Upon ligand binding it undergoes dimerization and autophosphorylation, activating downstream signalling pathways including the RAS-RAF-MEK-ERK and PI3K-AKT cascades. It is widely expressed across multiple human tissues, including skin, brain, colon, liver, and lung, with highest expressions observed in the placenta. EGFR has also been implicated as a component of the cytokine storm which contributes to severe form SARS-CoV-2 infections. Its role in human disease and in particular cancer has been well documented. Often it is associated with specific mutations that are prognostic and sometimes also causative to treatment failure. The most common activating mutations are deletions in exon 19 (del19 or E19del) and a point mutation called L858R in exon 211.
These mutations are critical targets for EGFR tyrosine kinase inhibitors (TKIs), which have significantly improved clinical outcomes for patients with EGFR mutant-driven cancers. More than 50% of acquired resistance to the first-generation EGFR-TKIs is caused by the T790M mutation, a threonine to methionine substitution at amino acid position 790 in exon 20 of the EGFR gene2-3. This mutation hampers the binding of first-generation reversible EGFR-TKIs to the kinase ATP-binding site of EGFR, leading to drug resistance. To overcome the secondary EGFR T790M mutation, third-generation EGFR-TKIs were developed. Osimertinib is an oral, highly potent, covalent third-generation EGFR-TKI that selectively targets EGFR-activating mutations as well as T790M resistance mutation4. Predictably however, resistance to covalent inhibitors has also emerged, in the form of the C797S mutation, which substitutes the reactive cysteine to a serine at amino acid position 797.
BI-4732 is a recently published5 novel fourth-generation EGFR-TKI, which is highly potent on the primary activating EGFR variants del19 and L858R and inhibits both variants also in the presence of the acquired EGFR resistance mutations T790M and/or C797S, while sparing wild-type EGFR. The compound is suitable for in vitro and in vivo studies, being orally bioavailable and demonstrating excellent CNS penetration properties5. Compared to the related inhibitor BI-8128, which is also available on opnMe, BI-4732 displays slightly higher potency in vitro, a longer residence time after iv dose in vivo, as well as better blood-brain barrier penetrating abilities.
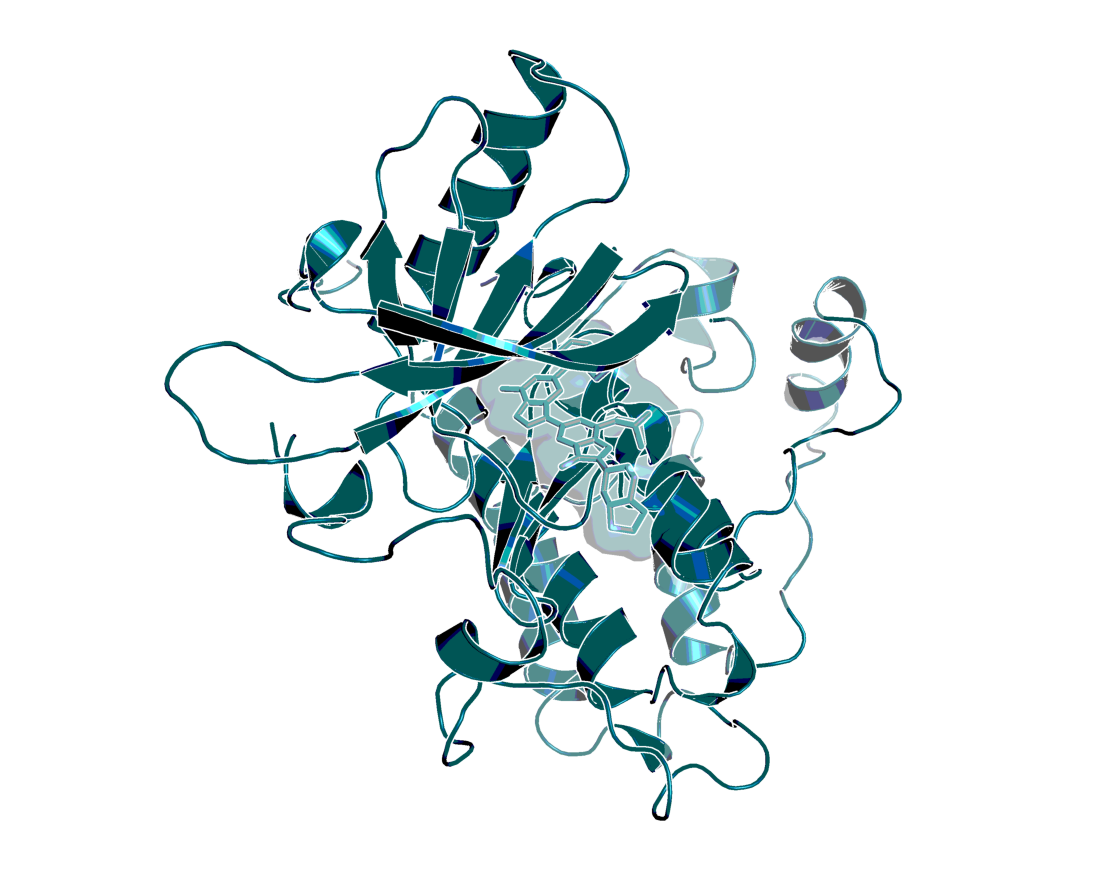
Model of the 3D structure of EGFR in complex with BI-4732, based on an in-house X-ray structure with a highly related inhibitor.
BI-4732 was profiled for potency and selectivity in engineered Ba/F3 and solid cancer cell lines in vitro (see table below for data). BI-4732 demonstrated potent, low nanomolar anti-proliferative activity in Ba/F3 cells that are dependent on the activity of human EGFR variants del19, del19 T790M C797S, L858R, and L858R T790M C797S. An EGFR-independent Ba/F3 cell line and a Ba/F3 clone dependent on EGFR wild-type signaling was only inhibited at much higher concentrations indicating low off-target cytotoxicity and sparing of EGFR wild-type activity. A parental human PC-9 NSCLC cell line (native EGFR del19 genotype) and a double resistance mutation containing engineered PC-9 T790M C797S variant (EGFR del19 T790M C797S) showed potent growth inhibition by BI-4732 in the single or low double digit nanomolar range, respectively. In line with the proliferation data, BI-4732 reduced the pharmacodynamic phospho-EGFR biomarker in the low nanomolar range in PC-9 variants with and without the resistance mutations T790M and C797S. BI-4732 inhibited A431 cell whose proliferation depends on EGFR wild-type signaling with markedly reduced potency compared to cell models harboring oncogenic EGFR variants demonstrating EGFR wild-sparing activity of the compound. The third-generation EGFR inhibitor and reference compound osimertinib showed severely impaired or no inhibitory activity in cell models harboring the EGFR C797S mutation, while potently inhibiting EGFR del19 and EGFR L858R cell models.
It should be noted that in a combination treatment with osimertinib, BI-4732 exhibited a synergistic effect in inhibiting PC-9 C797S (EGFR del19 C797S) cell proliferation, compared to monotherapy5.
| Probe name / REFERENCE COMPOUND | BI-4732 | BI-8128 | Osimertinib |
| MW [Da]a | 592.7 | 610.7 | 499.6 |
| Ba/F3 parental + IL-3 (EGFR-independent) (IC50) [nM]b | 1,367 | 3,558 | 1,008 |
| Ba/F3 EGFR wild-type + EGF ligand, proliferation, (IC50) [nM]b | 356 | 318 | 56 |
| Ba/F3 EGFR del19, proliferation, (IC50) [nM]b | 1.1 | 1.7 | 0.7 |
| Ba/F3 EGFR del19 T790M C797S, proliferation, (IC50) [nM]c | 2.6 | 3.3 | 635 |
| Ba/F3 EGFR L858R, proliferation, (IC50) [nM]c | 4.6 | 9.6 | 1.6 |
| Ba/F3 EGFR L858R T790M C797S, proliferation, (IC50) [nM]c | 7.8 | 16 | 588 |
| A549 (IC50) [nM]c | 1,064 | 1,615 | >1,000 |
| A431 (EGFR wild-type amplification), proliferation, (IC50) [nM]c | 730 | 492 | 112 |
| PC-9 parental (EGFR del19 genotype), proliferation, (IC50) [nM]c | 9 | 16 | n.d. |
| PC-9 T790M C797S (EGFR del19 T790M C797S genotype), proliferation, (IC50) [nM]c | 12 | 33 | >1,000 |
| PC-9 parental (EGFR del19 genotype), phospho-EGFR, (IC50) [nM]d | 6.5 | 9.6 | 3.3 |
| PC-9 T790M C797S (EGFR del19 T790M C797S genotype), phospho-EGFR, (IC50) [nM]d | 1.2 | 1.4 | >1,000 |
a The molecule is supplied in salt form. For the molecular weight of the salt form, please refer to the vial label
b Ba/F3 cell engineering and cellular proliferation assays were conducted according to the experimental protocol described in Reference 6.
c Solid cancer cell line engineering and cellular proliferation assays (A549, PC-9 variants and A431) were conducted according to the experimental protocol described in Reference 6.
d Phospho-EGFR inhibition assays were performed by treating the indicated cell lines with a concentration range of BI-4732, BI-8128 or osimertinib for 6 hours followed by cell lysis and phospho-EGFR quantification by semi-quantitative capillary western blotting.
BI-4732 displays good overall DMPK properties. Good solubility was identified at different pHs. In vitro hepatocyte stability data is in accordance with blood clearance data in animal models. Acceptable inhibition properties on a set of CYP-isoforms were also identified. Please note that BI-4732 may be less soluble compared to the related molecule BI-8128.
| Probe name | BI-4732 | BI-8128 |
| logD @ pH 11 | 3.6 | 3.4 |
| Solubility @ pH 7 [µg/mL] | <1 | 101 |
| MDCK permeability PappAB @ 1 µM [10-6 cm/s] | 9.1 | Low recovery |
| MDCK efflux ratio | 3.3 | n.a. |
| Microsomal stability (human/mouse/rat) [% QH] | 60 / 34 / 34 | 48 / <24.2 / 39 |
| Hepatocyte stability (human/mouse/rat) [% QH] | 27 / 45 / 40 | 56 / 31 / 22 |
| Plasma Protein Binding (human/mouse/rat) [%] | 98.4 / 99.3 / 98.5 | 97.6 / 98.1 / 96.0 |
| hERG [inh. % @ 1 µM] | 4.1 | 9.5 |
| hERG (IC50) [µM] | 14.3 | 5.6 |
| CYP 3A4 (IC50) [µM] | 2.7 | 6.1 |
| CYP 2C8 (IC50) [µM] | 40 | >50 |
| CYP 2C9 (IC50) [µM] | 13.0 | >50 |
| CYP 2C19 (IC50) [µM] | 16 | 30.6 |
| CYP 2D6 (IC50) [µM] | >50 | >50 |
For in vivo studies (oral administration) BI-4732 was formulated in 0.5% natrosol (acidified with HCl). For intravenous studies, the formulation included 25% HP-β-CD as well. BI-4732 shows low to moderate clearance, a medium volume of distribution and high bioavailability in animal models such as mouse and rat. Blood-brain barrier efflux assessed in rats revealed results superior brain permeability for BI-4732 (Kp,uu brain homogenate 0.7) compared to osimertinib (Kp,uu brain homogenate 0.21)5.
| BI-4732 | BI-8128 | |||
| Mousea | Ratb | Mousea | RATb | |
| Plasma Clearance [% QH] | 20.3 | 47.3 | 29 | 52 |
| Mean residence time after i.v. dose [h] | 10.3 | 8.5 | 2.5 | 3.8 |
| tmax [h] | 3 | 4 | 2 | 4 |
| Cmax [nM] | 747 | 252 | 1,200 | 437 |
| F [%] | 69 | 58 | 100 | 57 |
| Vss [L/kg] | 11.3 | 16.5 | 3.9 | 8.4 |
a i.v. dose: 1mg/kg; p.o. dose: 10 mg/kg
b i.v. dose: 1mg/kg; p.o. dose: 10 mg/kg
The in vivo efficacy of BI-4732 was investigated using the PC-9 cells subcutaneously implanted as xenograft models in mice. Oral administration of BI-4732 at a dose of 10 mg/kg bid and 25 mg/kg bi-daily (6 hours dosing interval between two daily doses) resulted in deep tumor regressions in a parental PC-9 xenograft model (EGFR del19 genotype) and in an engineered PC-9 T790M C797S xenograft model (EGFR del19 T790M C797S genotype).
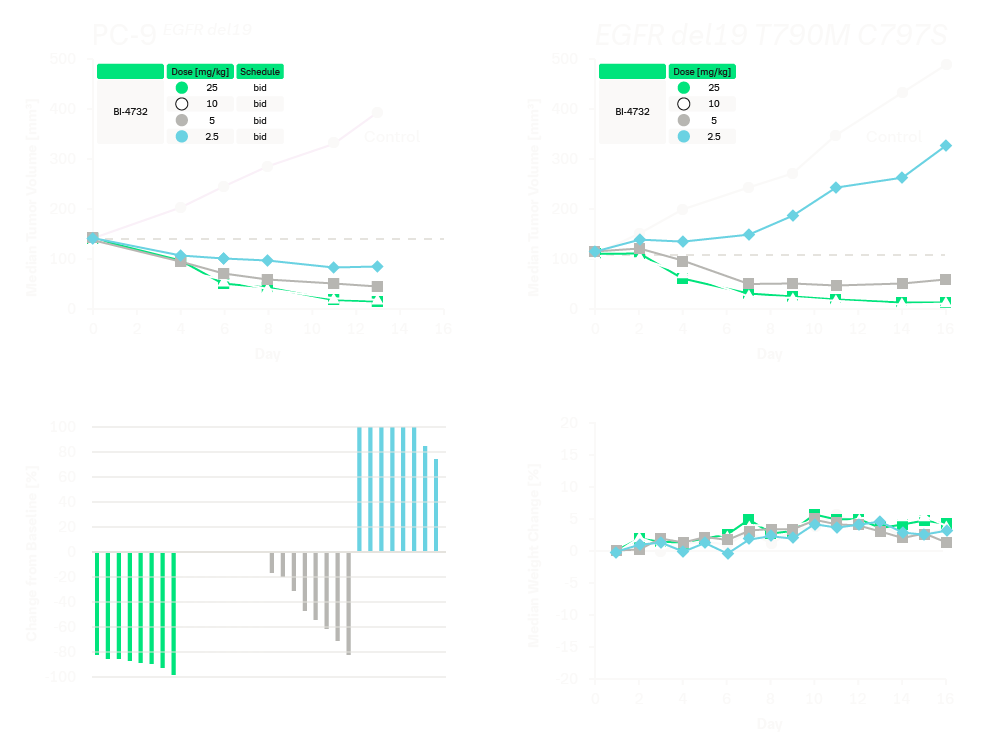
Oral administration of BI-4732 at a dose of 25 mg/kg bid induces tumor regressions in an isogenic set of PC-9 xenograft models with and without the resistance mutations T790M and C797S. Top panel: Median tumor volume post BI-4732 in subcutaneously implanted xenograft experiments employing an isogenic series of PC-9 NSCLC models with the indicated EGFR genotypes. Bottom panel: Waterfall plot representing the percentage of tumor volume change (left) and median body weight change (right) in mice with PC9 (EGFR del19 T790M C797S) xenografts following treatment with BI-4732.
The selectivity for EGFR was assessed in the SafetyScreen44™, where BI-4732 hit 7 out of 44 kinases more than 50% at the high concentration of 10 µM. BI-4732 was further tested in a kinase panel (number of kinases: 442) and showed selectivity for 340 targets (≤ 75% inhibition at a concentration of 1 µM). For comparison BI-8128 was tested on 205 targets in a selectivity panel and showed selectivity for 196 targets (≤ 50% inhibition @ 10 µM). In 13 assays (PDE4D2, M1/H, ALPHA1AH, M4/H, LCK_CE, HERG, M3/H, ADENOSINETRANSPORTER, PKCALPHA(H)@CE) BI-8128 showed inhibition between 51-100% @ 10µM.
| SELECTIVITY DATA AVILABLE | BI-4732 | BI-8128 |
| SafetyScreen44™ with kind support of | Yes | Yes |
| Invitrogen® | Yes | Yes |
| DiscoverX® | No | No |
Download selectivity data:
BI-4732_selectivityData.xlsx
BI-8128_selectivityData.xlsx
The third generation EGFR inhibitor osimertinib is commercially available.
BI-4732 is potent inhibitor against EGFR-activating mutations and on-target resistance mutations, with efficient activity in the central nervous system. It displays remarkable antitumor efficacy in vitro and in vivo as a single agent in various patient derived models with EGFRC797S-mediated osimertinib resistance.
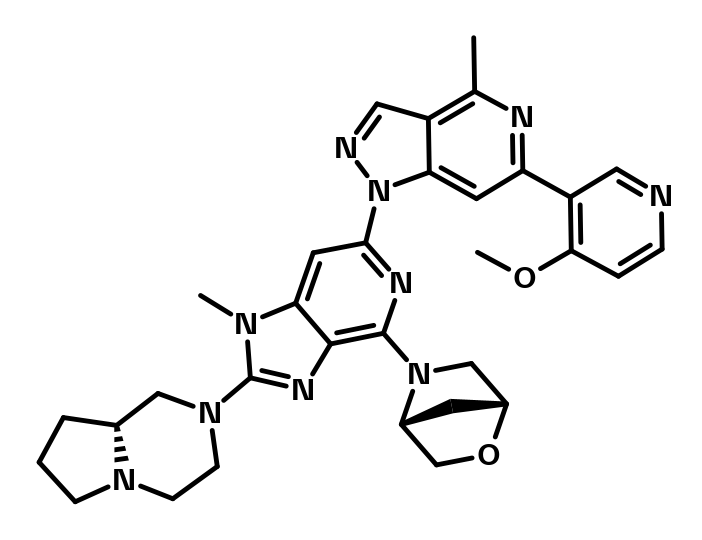
2 D structure formats available
{kind=link}
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma
Mok T. S., Wu Y., Thongprasert S., Yang C., Chu D., Saijo N., Sunpaweravong P., Han B., Margono B., Ichinose Y., Nishiwaki Y., Ohe Y., Yang J., Chewaskulyong B., Jiang H., Duffield E. L., Watkins C. L., Armour A. A., Fukuoka M.
N Engl J Med. 2009, 361(10):947-57.
AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer
Cross D. A. E., Ashton S. E., Ghiorghiu S., Eberlein C., Nebhan C. A., Spitzler P. J., Orme J. P., Finlay M. R. V., Ward R. A., Mellor M. J., Hughes G., Rahi A., Jacobs V. N., Red Brewer M., Ichihara E., Sun J., Jin H., Ballard P., Al-Kadhimi K., Rowlinson R., Klinowska T., Richmond G. H. P., Cantarini M., Kim D., Ranson M. R., Pao W.
Cancer Discov. 2014, 4(9):1046-61.
Discovery of a Novel Potent EGFR Inhibitor Against EGFR Activating Mutations and On-Target Resistance in NSCLC
Lee E. J., Oh S. Y., Lee Y. W., Kim J. Y., Kim M-J., Kim T. H., Lee J. B., Hong M. H., Lim S. M., Baum A., Woelflingseder L., Engelhardt H., Petronczki M., Solca F., Yun M. R., Cho B. C.
Clin Cancer Res. 2024, 30(8):1582-1594.
Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors
Engelhardt H., Böse D., Petronczki M., Scharn D., Bader G., Baum A., Bergner A., Chong E., Döbel S., Egger G., Engelhardt C., Ettmayer P., Fuchs J. E., Gerstberger T., Gonnella N., Grimm A., Grondal E., Haddad N., Hopfgartner B., Kousek R., Krawiec M., Kriz M., Lamarre L., Leung J., Mayer M., Patel N. D., Simov B. P., Reeves J. T., Schnitzer R., Schrenk A., Sharps B., Solca F., Stadtmüller H., Tan Z., Wunberg T., Zoephel A., McConnell D. B.
J Med Chem. 2019, 62(22):10272-10293.
When you plan a publication, please use the following acknowledgement:
BI-4732 was kindly provided by Boehringer Ingelheim via its open innovation platform opnMe, available at https://www.opnme.com.