KRAS G12C inhibitor
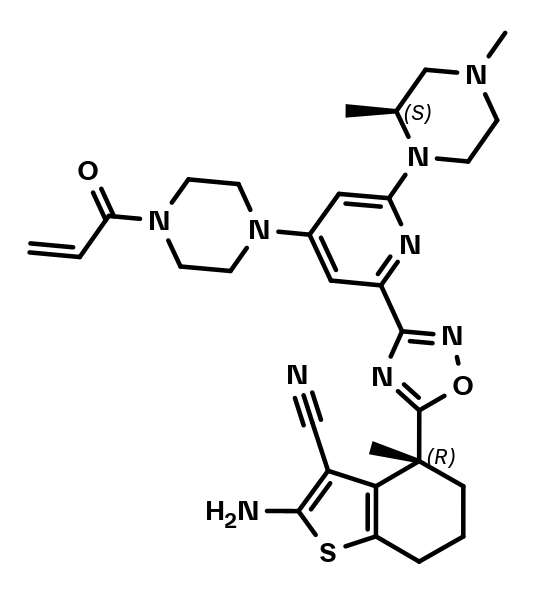
BI-0474
BI-0474 is an irreversible, covalent KRASG12C inhibitor showing high cellular activity and in vivo efficacy in an NCI-H358 xenograft model after intraperitoneal (ip) administration. BI-0474 has been discovered by Boehringer Ingelheim scientists in collaboration with the Fesik lab at Vanderbilt University, by optimizing non-covalent binding starting from a KRAS binder that was identified in an NMR-based fragment screen1,2.



More information
The KRAS oncoprotein is a GTPase acting as a key node in intracellular signaling pathways that are involved in cell growth and survival3,4. In normal cells, KRAS functions as a molecular switch, alternating between an inactive GDP-bound and an active GTP-bound state5. Transition between these states is modulated by guanine-nucleotide exchange factors (GEFs) such as SOS, and GTP hydrolysis, which is catalyzed by GTPase-activating proteins (GAPs), to inactivate KRAS. GTP-bound KRAS allows binding of effector proteins to trigger downstream signaling pathways, including the RAF-MEK-ERK (MAPK) pathway6-9. Activating mutations in KRAS are the most frequent oncogenic driver events in cancer10. Oncogenic KRAS mutations have been shown to shift the equilibrium between the two KRAS states towards the active, GTP-bound form. However, the major oncogenic versions of KRAS still undergo GAP-mediated GTP/GDP exchange and reactivation11. The oncogenic hotspot position 12, located at the lip of the switch II pocket, offers a covalent attachment point for KRASG12C inhibitors.
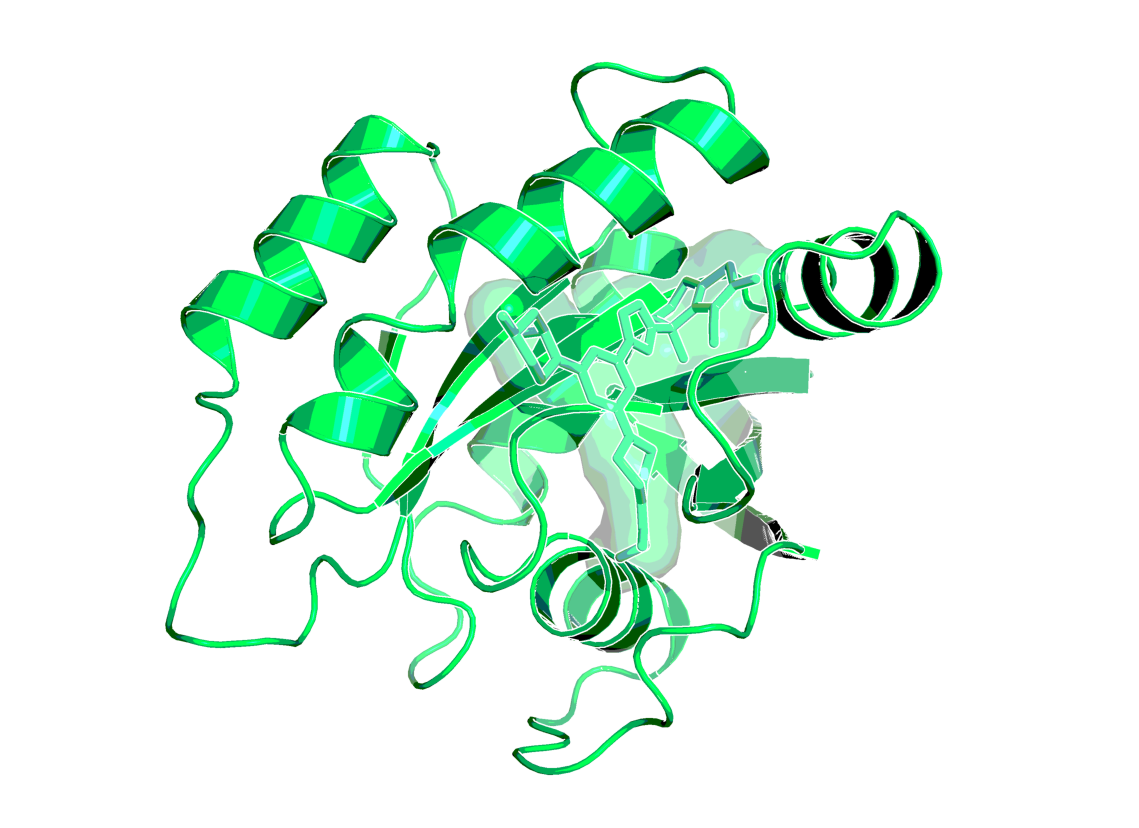
X-ray structure of the complex of BI-0474 with KRASG12C (PDB code 8AFB)1
BI-0474 shows single digit nM biochemical activity in a KRASG12C::SOS1 PPI assay and an antiproliferative effect at low nM concentrations in KRASG12C mutated cell lines like NCI-H358. As BI-0474 is a KRASG12C selective covalent inhibitor, it only has µM activity in the corresponding KRASG12D biochemical assay as well as µM antiproliferative activity against a KRASG12D mutated cell line. The corresponding negative control shows almost 200-fold lower biochemical activity in the KRASG12C::SOS1 assay.
| Probe name / Negative control | BI-0474 | BI-0473 |
| MW [Da, free base]a | 587.8 | 587.8 |
| KRASG12C::SOS1 AlphaScreen (IC50) [nM]b | 7 | 1,200 |
| KRASG12D::SOS1 AlphaScreen (IC50) [nM]b | 4,200 | 18,000 |
| EC50 NCI-H358 proliferation [nM]c | 26 | n.d. |
| EC50 GP2D proliferation [nM]d | 4,500 | n.d. |
a For the salt form you will get, please refer to the label on the vial and for the molecular weight of the salt, please refer to the FAQs
b BI-0474 is highly selective for KRASG12C compared to the KRASG12D mutant, as measured in vitro using an AlphaScreen assay. Details of the experimental procedure can be found in reference 1.
c, d BI-0474 shows potent antiproliferative activity on NCI-H358 cells carrying a G12C KRAS mutation. However, an antiproliferative effect on GP2D cells carrying a KRASG12D mutation was only observed in concentrations above 4 μM. Details of the experimental procedure can be found in reference 1.
BI-0474 shows medium predicted clearance and a high Caco-2 efflux ratio.
| Probe name / negative Control | BI-0474 | BI-0473 |
| logD @ pH 11 | 3.2 | 3.2 |
| Solubility @ pH 7 [µg/mL] | 99 | 2 |
| Caco-2 permeability AB @ pH 7.4 [*10-6 cm/s] | 0.8 | n.a. |
| Caco-2 efflux ratio | 45 | n.a. |
| MDCK permeability PappAB @ 1µM [10-6 cm/s] | 0.4 | n.a. |
| MDCK efflux ratio | 72 | n.a. |
| Microsomal stability (human/mouse/rat) [% QH] | 73 / 53 / 59 | 76 / 61 / 72 |
| Hepatocyte stability (human/mouse/rat) [% QH] | 40 / 14 / - | - / 15 / - |
| Plasma Protein Binding (human/mouse/rat) [%] | 96.7 / >98 / 98.3 |
|
| CYP 3A4 (IC50) [µM] | 15 | - |
| CYP 2C8 (IC50) [µM] | >50 | - |
| CYP 2C9 (IC50) [µM] | >50 | - |
| CYP 2C19 (IC50) [µM] | >50 | - |
| CYP 2D6 (IC50) [µM] | 21 | - |
BI-0474 shows decent exposure upon 25mg/kg po dosing as well as 50mg/kg ip dosing (higher ip doses not tested). Oral dosing of 100mg/kg did not lead to significantly increased exposure compared to 25mg/kg.
| BI-0474 | Mouse |
| Clearance [% QH]a | 20 |
| Mean residence time after i.v. dose [h] | 0.53 |
| tmax [h] @42.5µmol/kgb | 2 |
| Cmax [nM]b | 2,360 |
| F [%] | 23/24 |
| Vss [L/kg]a | 0.6 |
a i.v. dose mouse: 1 mg/kg (solution in 25% HP-b-CD, acidified with 0.1M HCl to pH 6)
b p.o. dose mouse: 25 mg/kg (suspension in Natrosol 0.5% or methyl cellulose)
c i.p. dose mouse: 50 mg/kg (solution in 25% HP-b-CD, acidified with 0.1M HCl to pH 6)
Target occupancy (TO) as well as pharmacodynamic (PD) biomarker modulation were assessed in the KRASG12C mutant NCI-H358 cell line-derived xenograft model after a daily treatment with 40 mg/kg ip on 3 consecutive days. As shown in Figure 4, MS-based TO analysis showed a strong and treatment-induced reduction of unmodified KRASG12C protein on the third day of consecutive treatment. This was confirmed by the analysis of RAS-GTP and p-ERK levels. Apoptosis induction was detectable in tumors in all 5 treated animals 6h post-dose on day 3, indicating that KRASG12C inhibition leads to induction of programmed cell death in this xenograft model. Anti-tumor efficacy was also assessed in this model, but as BI-0474 has not been optimized towards high oral bioavailability, it was administered ip once weekly or twice weekly on two consecutive days to explore whether infrequent systemic administration of covalent KRASG12C inhibitors could be efficacious. At 40 mg/kg BI-0474 showed anti-tumor efficacy in both dose groups with 68 % and 98 % tumor growth inhibition (TGI), respectively. Both dose groups showed some body weight loss, which is less pronounced for the low dose group compared to the high dose group.
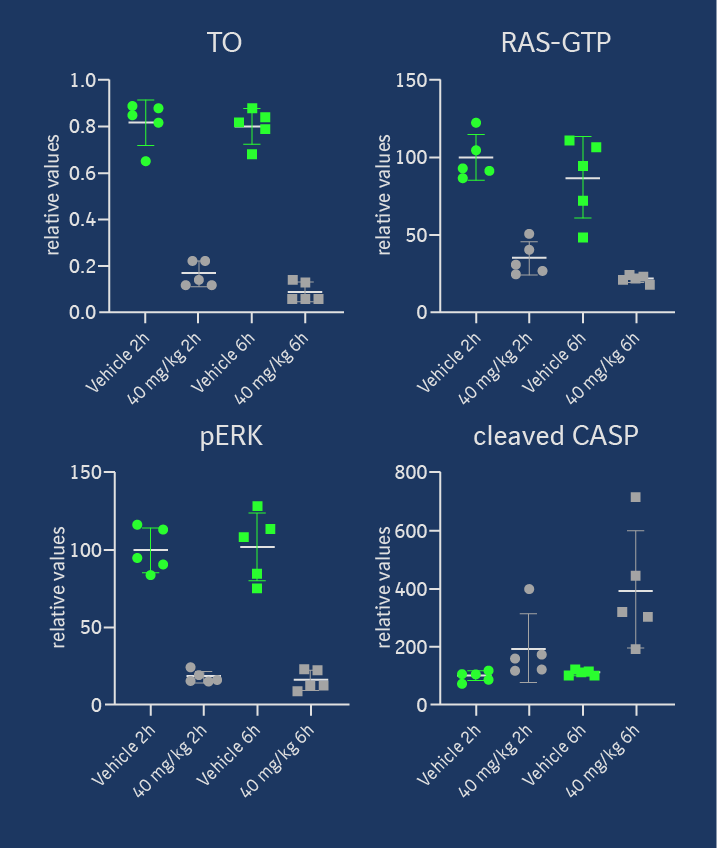
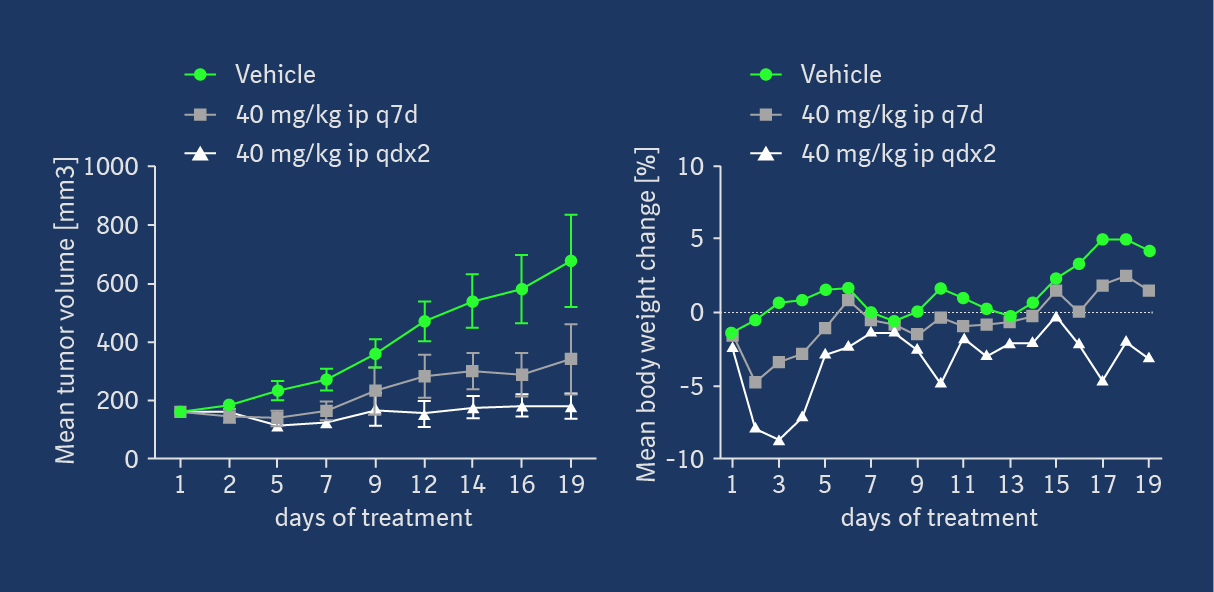
BI-0474 shows efficacy and PD biomarker modulation in an NCI-H358 cell line-derived non-small cell lung cancer xenograft model; left: treatment with BI-0474 leads to TO (decrease of unmodified KRASG12C protein levels measured by MS), reduction of RAS-GTP and pERK levels, as well as apoptosis induction in vivo; right: BI-0474 shows efficacy in NCI-H358 xenograft (mean tumor volumes and mean body weight change shown vs. control (green circles), 40 mg/kg BI-0474 ip once/week (q7d, grey squares), 40 mg/kg BI-0474 ip twice/week on 2 consecutive days (qdx2, white triangles)); Details of the experimental procedures can be found in reference 1.
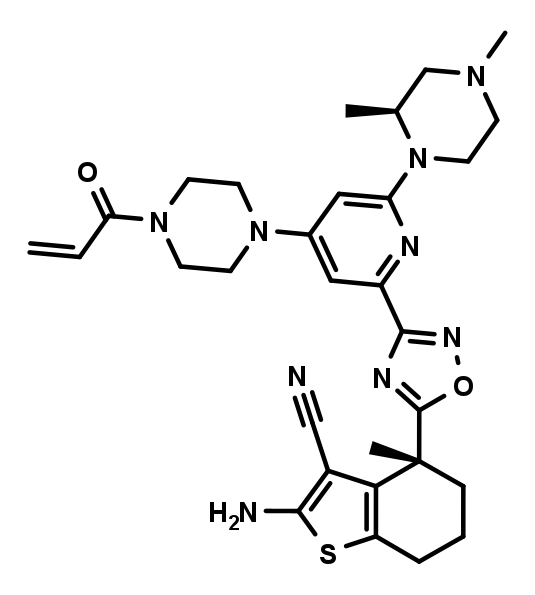
The negative control BI-0473 is a diastereomer of BI-0474 and also carries an electrophilic acrylamide warhead. However due to the inverted stereo center close to the amino-cyano-thiophene, which serves as one of the key pharmacophores, the non-covalent binding contribution is significantly reduced, resulting in a 200-fold lower biochemical activity compared to BI-0474.
BI-0473 which serves as a negative control
As expected for a covalent KRASG12C inhibitor the compound shows a strong selectivity for KRASG12C vs other mutations (see KRASG12C vs. KRASG12D AlphaScreen data in the provided in vitro activity table). In the SafetyScreen44TM at a high concentration of 10 µM the compound hit 9 of 44 targets (M3/H, ALPHA1AH, COX-2, Ca+, MU/H, M2/H, COX-1, DATRANS, 5HT2B). Negative control BI-0473 hit 6 of 44 targets in SafetyScreen44TM at a high concentration of 10 µM (COX-2, KAPPA, 5HT2N, M2, COX-1, 5HT1B).
| Selectivity data available | BI-0474 | BI-0473 |
| SafetyScreen44™ with kind support of | Yes | Yes |
| Invitrogen® | No | No |
| DiscoverX® | No | No |
| Dundee | No | No |
Download selectivity data:
BI-0474_selectivityData.xlsx
BI-0473_selectivityData.xlsx
BI-0474 is a covalent KRASG12C inhibitor showing high cellular activity and in vivo efficacy after intraperitoneal administration. It was discovered using structure-based design approaches, by optimizing non-covalent binding of a fragment hit1.
{kind=link}
{kind=link}
Fragment Optimization of Reversible Binding to the Switch II Pocket on KRAS Leads to a Potent, In Vivo Active KRASG12C Inhibitor
Broeker J., Waterson A. G., Smethurst C., Kessler D., Böttcher J., Mayer M., Gmaschitz G., Phan J., Little A., Abbott J. R., Sun Q., Gmachl M., Rudolph D., Arnhof H., Rumpel K., Savarese F., Gerstberger T., Mischerikow N., Treu M., Herdeis L., Wunberg T., Gollner A., Weinstabl H., Mantoulidis A., Kraesaidmer O., McConnell D. B., Fesik S. W.
J Med Chem 2022, 65 (21), 14614–14629.
When you plan a publication, please use the following acknowledgement:
BI-0474 was kindly provided by Boehringer Ingelheim via its open innovation platform opnMe, available at https://www.opnme.com.