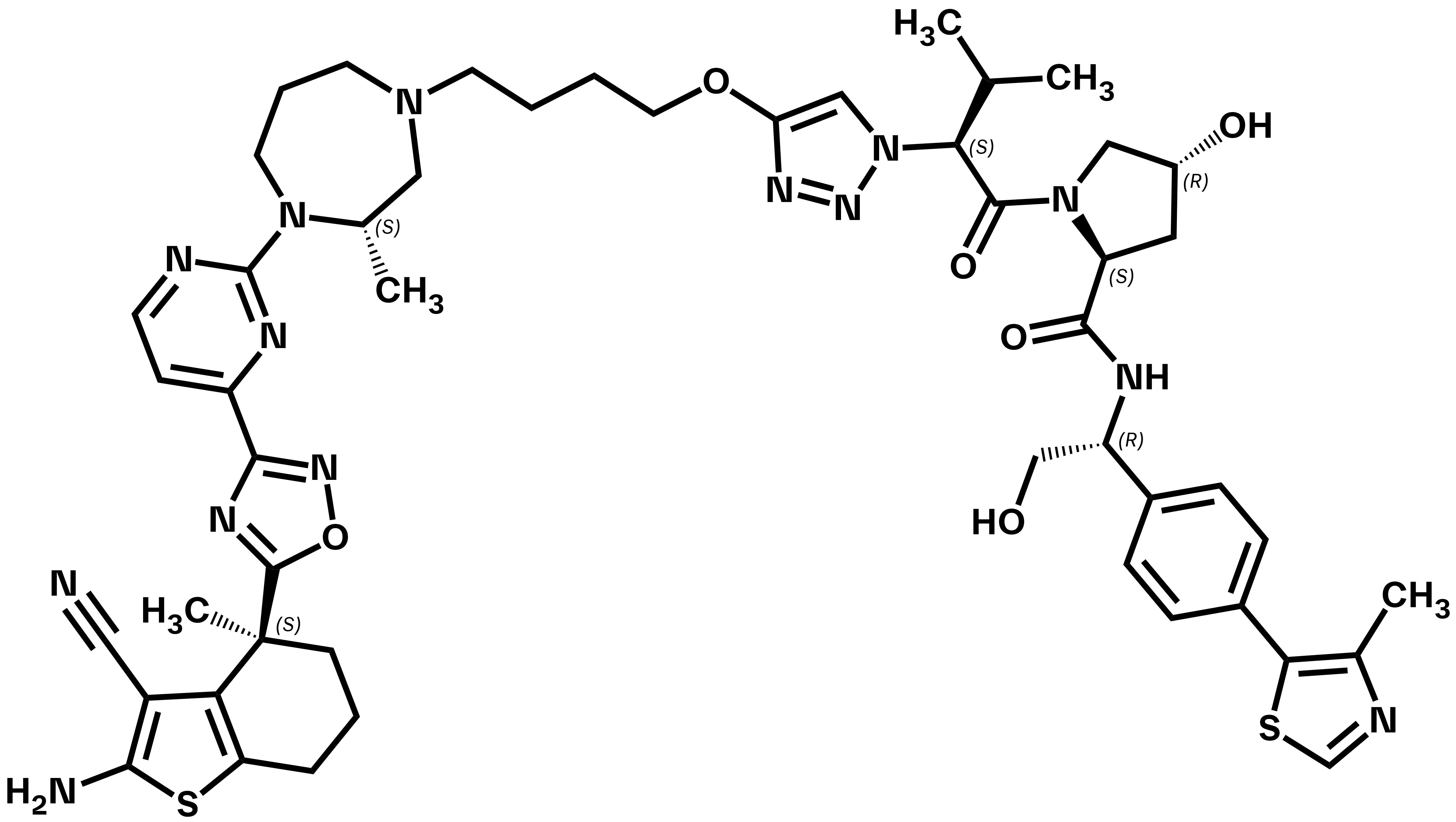
pan-KRAS VHL PROTAC
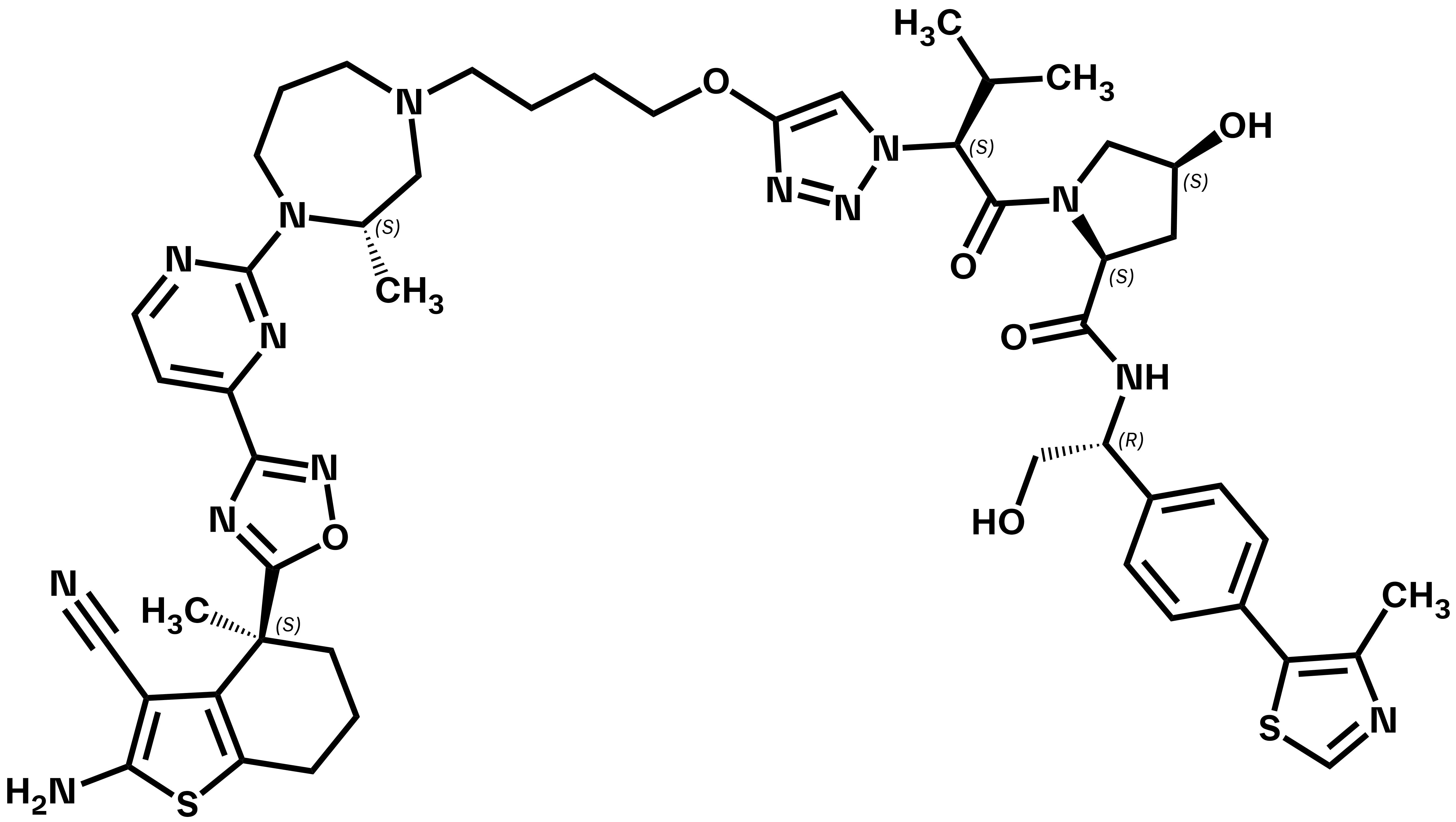
ACBI3
ACBI3 is a selective and potent pan-KRAS degrader discovered via a structure-based design approach guided by optimization of VHL:PROTAC:KRAS ternary complex stability and durability1,2. It acts on the majority of KRAS mutants and inhibits proliferation in KRAS mutant cell lines covering a wide range of tumor types. ACBI3 suppresses oncogenic KRAS protein levels in vivo, leading to potent and long-lasting reduction of KRAS signalling1.




More information
Kirsten rat sarcoma viral oncogene homologue (KRAS) is the most commonly mutated oncogene in human cancers. Variants, predominantly mutations at glycine (G) 12 or glutamine (Q) 61, increase the proportion of activated, GTP-loaded KRAS, enhancing RAF-MEK-ERK (MAPK) signaling, and drive tumor growth3. Developed in collaboration with Alessio Ciulli (Centre for Targeted Protein Degradation, University of Dundee), ACBI3 is a first in class proteolysis targeting chimera (PROTAC) potently degrading 13 out of 17 of the most prevalent KRAS variants (G12A, G12C, G12D, G12V, G13C, G13D, G13V, Q61E, Q61H, Q61P, A146P, A146T, and A146V), while sparing the closely related paralogues H- and NRAS. ACBI3 is poorly soluble and lacks oral bioavailability but can be administered parenterally for in vivo experiments. A matched negative control, cis-ACBI3, is available.
Compared with inhibition, degradation of KRAS results in more profound and sustained pathway modulation across a broad range of KRAS mutant cell lines. As a result, ACBI3 inhibits growth of the majority of cancer cell lines driven by KRAS mutations while sparing models without genetic KRAS aberrations. Adequately formulated4, ACBI3 achieves degradation of KRAS leading to tumor regression in select models in vivo.

X-ray structure of the complex of ACBI3 with KRAS (PDB code 8qvu)1.
ACBI3 exhibits potent intracellular VHL engagement, ternary complex formation, and ubiquitination, translating into efficient E3-ligase dependent cellular degradation and proteome-wide selectivity. ACBI3 displays antiproliferative activity in a cell line panel on KRAS mutant but not KRASWT cell lines (geometric mean IC50 = 478 nM vs 8.3 µM, respectively).
| Probe name / Negative control | ACBI3 | cis-ACBI3 |
| MW [Da]a | 1,019.25 | 1,019.25 |
| Cellular KRASG12D degradation, 24 h (GP5d cells) (DC50) [nM]b | 2 | >1,000 |
| Cellular KRASG12V degradation, 24 h (SW620 cells) (DC50) [nM]b | 7 | n.d. |
| Cellular proliferation, 5 days (GP5d cells) (IC50) [nM]c | 5 | >1,000 |
| Cellular proliferation, 5 days (SW620 cells) (IC50) [nM]c | 15 | n.d. |
aPlease note that ACBI3 and cis-ACBI3 are supplied in salt form; for the molecular weight of the salt, please refer to the vial label
bby capillary electrophoresis using the following antibodies: KRASG12D (Cell Signaling #14429), KRASG12V (Cell Signaling #14412), normalized by GAPDH (Abcam #ab9485),
cby CellTiterGlo assay (Promega #G7570)
ACBI3 is a large, lipophilic molecule with low aqueous solubility at neutral pH. It has low stability in liver microsomes while being good to moderately stable in hepatocytes. ACBI3 displays high plasma protein binding and has a low absorptive permeability with a high efflux ratio as determined by the Caco2-assay.
| Probe name / Negative control | ACBI3 | cis-ACBI3 |
| logD @ pH 11 | 4.06 | 4.27 |
| Solubility @ pH 6.8 [µg/ml] | <1 | <1 |
| Caco-2 permeability AB [*10-6 cm/s] | 0.8 | n.d. |
| Caco-2 efflux ratio | 20 | n.d. |
| Microsomal stability (human/mouse/rat) [% QH] | >88/>88/84 | >88/>88/75 |
| Hepatocyte stability (human/mouse/rat) [% QH] | <24/46/68 | n.d. |
| Plasma Protein Binding (human/mouse/rat) [%] | >99.74/99.95/>99.74 | n.d. |
ACBI3 displays moderate clearance in mice after intravenous dosing and a favorable volume of distribution. When formulating ACBI3 in PEG-400/Transcutol/Kolliphor HS 15 to enable subcutaneous dosing, a Cmax of 70 nM and a tmax of 2 h are reached.
No in vivo DMPK parameters were measured for negative control cis-ACBI3.
| ACBI3 | Mouse |
| Clearance mL/(L·h)]a | 39 |
| Mean residence time after i.v. dose [h]a | 0.67 |
| tmax [h]b | 2 |
| Cmax [nM]b | 70 |
| Vss [l/kg]a | 1.6 |
a i.v. dose 2 [mg/kg] in HPβCD
b s.c. dose 30 [mg/kg] in PEG-300/Transcutol/Solutol 60/30/10 (v/v/v)
ACBI3 lacks oral bioavailability. Therefore, tumor-bearing mice were intraperitoneally dosed with a nano-milled suspension of ACBI3 (ZentriMix 380R dual centrifuge, Andreas Hettich GmbH & Co. KG, zirconium oxide milling beads, 4 hours, 1,000 rpm, 4 °C) formulated in hydroxypropyl cellulose, polysorbate 80 and SDS at an injection volume of 5 mL/kg. ACBI3 dosed at 30 mg/kg daily induced regressions in mice engrafted with RKN cells which depend on KRASG12V. Other formulations and parenteral routes of administration (e.g., subcutaneous delivery) were tested and found not to be suitable for efficacy studies due to local intolerability.
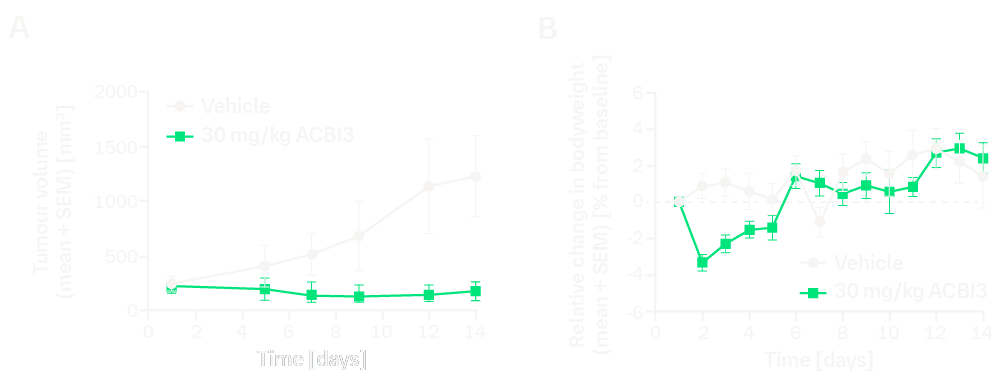
In vivo efficacy of ACBI3 in the RKN xenograft model, administered intraperitoneally daily, formulated as a nano-milled suspension (N=7). (A) Tumor volume, (B) relative change in body weight. Timescale starts at the day of randomization.
Cis-ACBI3 is a stereoisomer of ACBI3 displaying deficient binding to VHL, and hence can be used as a negative control for KRAS degradation. Note, however, that cis-ACBI3 still binds KRAS and acts as a non-covalent KRAS inhibitor.
cis-ACBI3, which serves as a negative control.
The degradation selectivity of ACBI3 was assessed by whole cell proteomics MS analysis of GP2d cells treated with ACBI3 or the inactive stereoisomer cis-ACBI3. The below figure validates selective degradation of KRAS. Of note, HRAS (log2 fold change -0.0006, -logP 0.001) and NRAS (log2 fold change -0.12, -logP 0.52) levels are not significantly affected.
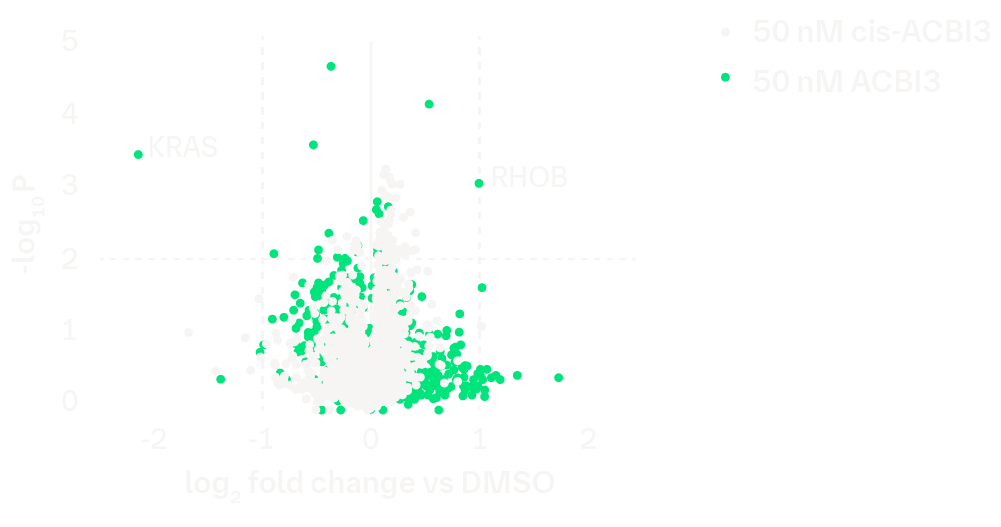
Volcano plot of whole cell proteomics MS analysis of GP2d cells treated with 50 nM ACBI3 or inactive stereoisomer cis-ACBI3 (8 hours, N=3).
ACBI3 is a first in class VHL-recruiting PROTAC molecule, potently degrading multiple KRAS variants in vitro and in vivo. Proteolytic degradation of KRAS via ACBI3 enables a greater than 10-fold higher potency compared to target inhibition and results in prolonged suppression of MAPK signaling.
{kind=link}
{kind=link}
Targeting cancer with small molecule pan-KRAS degraders
Popow J., Farnaby W., Gollner A., Kofink C., Fischer G., Wurm M., Zollman D., Wijaya A., Mischerikow N., Hasenoehrl C., Prokofeva P., Arnhof H., Arce-Solano S., Bell S., Boeck G., Diers E., Frost A.B., Goodwin-Tindall J., Karolyi-Oezguer J., Khan S., Klawatsch T., Koegl M., Kousek R., Kratochvil B., Kropatsch K., Lauber A.A., McLennan R., Olt S., Peter D., Petermann O., Roessler V., Stolt-Bergner P., Strack P., Strauss E., Trainor N., Vetma V., Whitworth C., Zhong S., Quant J., Weinstabl H., Kuster B., Ettmayer P., Ciulli A.
Science 2024, 385(6715), 1338-1347.
Catalytic in vivo protein knockdown by small-molecule PROTACs
Bondeson D. P., Mares A., Smith I. E. D., Ko E., Campos S., Miah A. H., Mulholland K. E., Routly N., Buckley D. L., Gustafson J. L., Zinn N., Grandi P., Shimamura S., Bergamini G., Faelth-Savitski M., Bantscheff M., Cox C., Gordon D. A., Willard R. R., Flanagan J. J., Casillas L. N., Votta B. J., den Besten W., Famm K., Kruidenier L., Carter P. S., Harling J. D., Churcher I., Crews C. M.
Nat. Chem. Biol. 2015, 11, 611–617.
When you plan a publication, please use the following acknowledgement:
ACBI3 was kindly provided by Boehringer Ingelheim via its open innovation platform opnMe, available at https://www.opnme.com.