mGluR1 Positive Allosteric Modulators
BI02982816
BI02982816 and BI-1752 are positive allosteric modulators of the metabotropic glutamate receptor subtype 1 (mGluR1), suitable for in vivo studies. BI-1752, the chemically optimized reiteration of BI02982816, features improved drug metabolism and pharmacokinetic properties while maintaining efficacy preclinically. The molecules emerged from a collaboration with the Warren Center for Neuroscience Drug Discovery at Vanderbilt University.





More information
mGluR1 receptors are Gq/11-coupled metabotropic glutamate receptors predominantly expressed in the central nervous system, particularly within the cortico-striatal and thalamo-striatal circuits. Activation of mGluR1 enhances intracellular calcium signaling and promotes endocannabinoid (eCB) release, which in turn inhibits dopamine (DA) release via CB2 receptor activation on presynaptic terminals1. This mechanism is especially relevant in the dorsolateral striatum, a region implicated in the pathophysiology of schizophrenia1.
Positive allosteric modulators of mGluR1 potentiate this signaling cascade, leading to normalization of hyperdopaminergic states in preclinical models without directly inhibiting D1 receptor signaling. Preclinical evidence indicates that this may preserve motivational and cognitive functions1-4.
In addition, mGluR1 PAMs have demonstrated antipsychotic-like efficacy in preclinical models, including reversal of amphetamine-induced hyperlocomotion and MK-801-induced cognitive deficits 1,5,6,7. Genetic studies have linked GRM1 mutations to schizophrenia, and mGluR1 PAMs have shown the ability to restore function in mutant receptors8. Together, these findings suggest a potential role of mGluR1 PAMs in impacting positive and cognitive symptoms of schizophrenia through modulation of glutamatergic and dopaminergic signaling in disease-relevant brain circuits1-9.
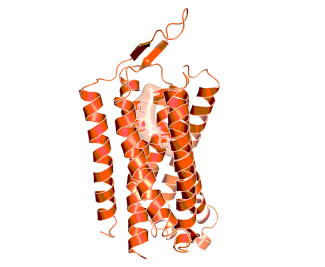
Transmembrane domain of mGluR1 in complex with a negative allosteric modulator, indicating the location of the allosteric binding site, as observed by X-ray crystallography (PDB code: 4or2.pdb)10.
The potency of mGluR1 PAMs was determined using calcium flux measurements, as described in previous studies7,11. From a weak high-throughput screening hit (EC50 >10 µM, 71% Glumax), optimization efforts improved functional potency over 350-fold to deliver the selective (inactive on mGlu2-5,7,8) and CNS penetrant (rat Kp = 0.99, Kp,uu = 0.82; MDCK-MDR1 ER = 1.7, Papp = 73 x 10-6 cm/s) mGluR1 PAM (BI02982816, EC50 = 54 nM, 83% Glumax).
| Probe names / Negative control | BI02982816 | BI-1752 | BI-4066 |
| MW [Da]a | 396 | 367 | 426 |
| Human mGluR1 (EC50) [nM]b/Emax [%]b | 54 / 83 | 39 / 65 | > 10,000 / 42 |
| Rat mGluR1 (EC50) [nM]b/Emax [%] | 46 / 124 | 107 / 94 | n.a. |
a For the salt form you will get, please refer to the label on the vial and for the molecular weight of the salt, please refer to the FAQs
b assay conditions – Calcium mobilization assay, see reference 8
BI02982816 has an acceptable fraction unbound in plasma while predicted hepatic clearance is moderate across species. BI02982816 is not a human P-gp substrate, but has an acceptable CYP450 profile, and shows no 3A4 mechanism-based inhibition.
BI-1752 has an advanced in vitro DMPK profile with acceptable hepatic clearance across species and good fraction unbound in plasma. Compared to BI02982816, this compound has an improved CYP450 profile and is predicted to be highly CNS penetrant in humans and is found to be highly CNS penetrant in rats, respectively.
Finally, the negative control BI-4066 shows moderate predicted brain efflux and stability across species, while demonstrating an excellent CYP450 profile.
| Probe names / Negative control | BI02982816 | BI-1752 | BI-4066 |
| logD @ pH 11/2 | 2.7 / 2.6 | 3.3 | 1.6 / 0.2 |
| Solubility @ pH 7 [µg/mL] | < 0.001 | < 1 | >103 |
| MDCK permeability PappAB @ 1µM [10-6 cm/s] | 35.0 | 41.0 | 34 |
| MDCK efflux ratio | 0.7 | 0.9 | 2.5 |
| Microsomal stability (human/mouse/rat) [% QH] | 81 / 82 / 51 | 29 / n.a. / 42 | 52 / 62 / 31 |
| Hepatocyte stability 50% (human/mouse/rat) [% QH] | 29 / 96 / 11 | 13 / 80 / 32 | n.a. |
| Plasma Protein Binding (human/mouse/rat) [%] | 98.8 / 98 / 98 | 95.7 / 96 / 94 | n.a. |
| hERG (IC50) [µM] | > 10.0 | 3.5 | n.a. |
| CYP 3A4 (IC50) [µM] (MDZ) | 50.0 | 50.0 | >50 |
| CYP 2C8 (IC50) [µM] | 3.9 | 45.6 | 43.5 |
| CYP 2C9 (IC50) [µM] | 21.4 | 50.0 | >50 |
| CYP 2C19 (IC50) [µM] | 25.6 | 26.8 | >50 |
| CYP 2D6 (IC50) [µM] | 50.0 | 50.0 | >50 |
| CYP 1A2 (IC50) [µM] | 43.5 | 48.0 | >50 |
| CYP 2B6 (IC50) [µM] | 20.7 | 50.0 | >50 |
BI-1752, the chemically optimized iteration of BI02982816, features improved drug metabolism and pharmacokinetic properties while maintaining efficacy in preclinical in vivo models. Both BI02982816 and BI-1752 demonstrate good brain exposure at their respective minimum effective oral doses in rats: 3 mg/kg for BI02982816 yielding a free brain concentration of 32 nM (~0.6× EC50), and 10 mg/kg for BI-1752 yielding 80 nM (~0.74× EC50). Dose escalation to 10 mg/kg for BI02982816 increased exposure to 67 nM (~1.2× EC50) without adverse effects, while 30 mg/kg of BI-1752 raised free brain levels to ~579 nM (~5.2× EC50) but was associated with adverse events, limiting further dose escalation.
| Probe name / Negative control | BI02982816 | BI-1752 |
| Clearance (rat/mouse) [% QH]a | 11.9 / n.a. | 65.5 / 102 |
| Mean residence time after i.v. dose (rat/mouse) [h]a | 3.5 / n.a. | 1.2 / 0.3 |
| tmax [h]b | 2 h | 0.5 |
| Cmax [nM]b | 10,500 | 2,400 |
| F [%]b | 100 | 42.8 |
| Vss (rat/mouse) [L/kg]a | 1.8 / n.a. | 3.3 / 1.8 |
a i.v. dose: rat= 0.58 mg/kg, mouse= 0.58 mg/kg
b p.o. dose: rat= 3.0 mg/kg, mouse= 5.8mg/kg
BI02982816 was active in amphetamine-induced hyperlocomotion (minimum effective dose (MED) = 3 mg/kg, p.o.) and MK-801 (antagonist of the NMDA receptor) induced disruptions of novel object recognition (MED = 10 mg/kg p.o.). Similarly, BI-1752 showed activity in both models. It reduced amphetamine-induced hyperlocomotion (MED = 3 mg/kg p.o.) and improved novel object recognition (NOR) disrupted by MK-801 (MED = 10 mg/kg p.o.). Furthermore, BI-1752 exhibited a clear pharmacokinetic–pharmacodynamic (PK/PD) relationship.
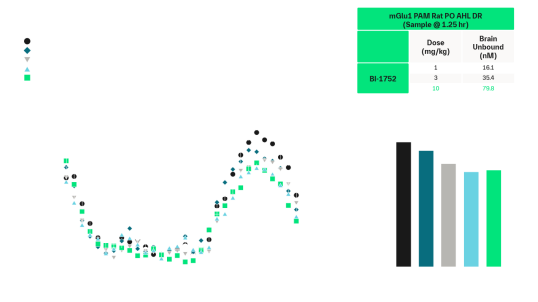
Rat amphetamine-induced hyperlocomotion and reversal by mGluR1 PAMs. Amphetamine (0.75 mg/kg s.c.) induced robust hyperlocomotion, which was dose-dependently reversed by oral administration (0.5% Natrasol/0.015% Tween 80) of BI-1752. A clear PK/PD relationship with efficacy was noted at ~0.7-fold the in vitro rat mGluR1 EC50 in unbound brain, in agreement with BI0298281611.
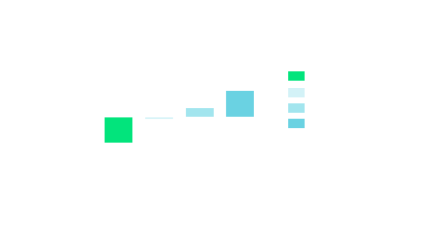
Rat MK-801 (NMDA receptor antagonist) induced disruption of novel object recognition (NOR) and reversal by BI02982816. MK-801 (0.5 mg/kg i.p.) induced a robust disruption of NOR, which was dose-dependently reversed by oral administration (10% tween 80) of BI029828167.
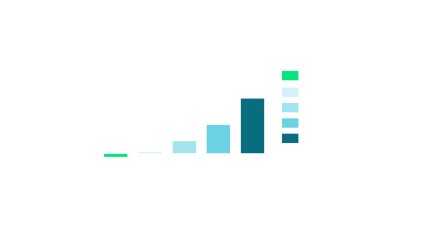
Rat MK-801-induced disruption of novel object recognition and reversal by BI-1752. MK-801 (0.075 mg/kg SC) induced a robust disruption of NOR, which was dose-dependently reversed by oral administration (0.5% Natrasol/0.015% Tween 80 in water). N = 12−15/group of male Sprague−Dawley rats. One-way ANOVA: p = 0.1311.
BI-4066, a structurally close analog of BI-1752, can be used as negative control.
BI-4066 which serves as a negative control
Both BI02982816 and BI-1752 are highly selective versus mGluR2-8 as well as against a broad panel of GPCRs, ion channels, and transporters. BI02982816 inhibited 5HT2B/H with 70% inhibition at 10mM. BI-1752 and also the negative control BI-4066 inhibited none of the targets with > 50% at a 10μM in the SafetyScreen44™.
| SELECTIVITY DATA AVILABLE | BI02982816 | BI‑1752 | BI‑4066 |
| SafetyScreen44™ with kind support of | Yes | Yes | Yes |
Download selectivity data:
BI-2816_selectivityData.xlsx
BI-1752_selectivityData.xlsx
BI-4066_selectivityData.xlsx
Ro 07-11401 and VU6004909 are tool compounds for in vivo target validation studies1.
BI02982816 and its chemically optimized reiteration BI-1752 are positive allosteric modulators of metabotropic glutamate receptors that play a role in the regulation of neurotransmission and synaptic plasticity. BI-4066 is available as negative control.
2D structure formats available
mGluR1 Positive Allosteric Modulators | BI029816.png
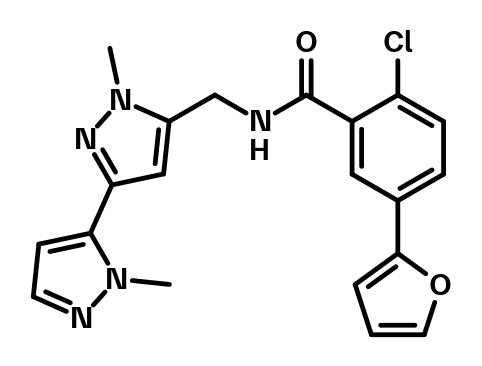{kind=link}
mGluR1 Positive Allosteric Modulators | BI029816.smiles
mGluR1 Positive Allosteric Modulators | BI029816.sdf
mGluR1 Positive Allosteric Modulators | BI-1752.png
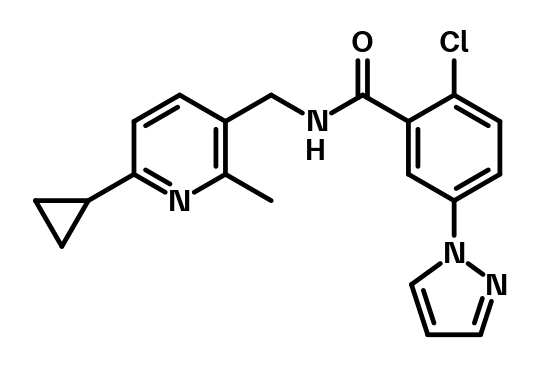{kind=link}
mGluR1 Positive Allosteric Modulators | BI-1752.smiles
mGluR1 Positive Allosteric Modulators | BI-1752.sdf
Negative control | BI-4066.png
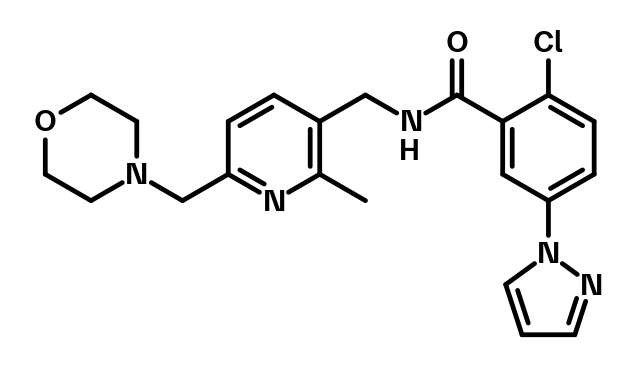{kind=link}
Activation of the mGlu1 metabotropic glutamate receptor has antipsychotic-like effects and is required for efficacy of M4 muscarinic receptor allosteric modulators
Yohn S. E., Foster D. J., Covey D. P., Moehle M. S., Galbraith J., Garcia-Barrantes P. M., Cho H. P., Bubser M., Blobaum A. L., Joffe M. E., Cheer J. F., Jones C. K., Lindsley C. W., Conn P. J.
Mol Psychiatry 2020, 25(11), 2786–2799.
Development of Novel, CNS Penetrant Positive Allosteric Modulators for the Metabotropic Glutamate Receptor Subtype 1 (mGlu1), Based on an N-(3-Chloro-4-(1,3-dioxoisoindolin-2-yl)phenyl)-3-methylfuran-2-carboxamide Scaffold
Garcia-Barrantes P. M., Cho H. P., Niswender C. M., Byers F. W., Locuson C. W., Blobaum A. L., Xiang Z., Rook J. M., Conn P. J., Lindsley C. W.
J Med Chem 2015, 58(20), 7959–7971.
Lead optimization of the VU0486321 series of mGlu(1) PAMs. Part 2: Sar of alternative 3-methyl heterocycles and progress towards an in vivo tool
Garcia-Barrantes P. M., Cho H. P., Metts A. M., Blobaum A. L., Niswender C. M., Conn P. J., Lindsley C. W.
Bioorg Med Chem Lett 2016, 26(3), 751–756.
Mglu1 potentiation enhances prelimbic somatostatin interneuron activity to rescue schizophrenia-like physiological and cognitive deficits
Maksymetz J., Byun N. E., Luessen D. J., Li B., Barry R. L., Gore J. C., Niswender C. M., Lindsley C. W., Joffe M. E., Conn P. J.
Cell Rep 2021, 37(5), 109950.
Mglu1-mediated restoration of prefrontal cortex inhibitory signaling reverses social and cognitive deficits in an NMDA hypofunction model in mice
Luessen D. J., Gallinger I. M., Ferranti A. S., Foster D. J., Melancon B. J., Lindsley C. W., Niswender C. M., Conn P. J.
Neuropsychopharmacology 2022, 47(10), 1826–1835.
Discovery of VU6024578/BI02982816: An mGlu1 Positive Allosteric Modulator with Efficacy in Preclinical Antipsychotic and Cognition Models
Reed C. W., Kalbfleisch J. F., Turkett J. A., Trombley T. A., Nastase A. F., Spearing P. K., Haymer D. H., Sarwar M. M., Quitalig M., Dickerson J. W., Blobaum A. L., Boutaud O., Voehringer P., Schuelert N., Cho H. P., Niswender C. M., Rook J. M., Priepke H., Ursu D., Conn P. J., Melancon B. J., Lindsley C. W.
J Med Chem 2024, 67(24), 22291–22312.
Chemical modulation of mutant mGlu1 receptors derived from deleterious GRM1 mutations found in schizophrenics
Cho H. P., Garcia-Barrantes P. M., Brogan J. T., Hopkins C. R., Niswender C. M., Rodriguez A. L., Venable D. F., Morrison R. D., Bubser M., Daniels J. S., Jones C. K., Conn P. J., Lindsley C. W.
ACS Chem Biol 2014, 9(10), 2334–2346.
Antipsychotic-like Effects of M4 Positive Allosteric Modulators Are Mediated by CB2 Receptor-Dependent Inhibition of Dopamine Release
Foster D. J., Wilson J. M., Remke D. H., Mahmood M. S., Uddin M. J., Wess J., Patel S., Marnett L. J., Niswender C. M., Jones C. K., Xiang Z., Lindsley C. W., Rook J. M., Conn P. J.
Neuron 2016, 91(6), 1244–1252.
Human class C G protein-coupled metabotropic glutamate receptor 1 in complex with a negative allosteric modulator
Wu H., Wang C., Gregory K. J., Han G. W., Cho H. P., Xia Y., Niswender C. M., Katritch V., Cherezov V., Conn P. J., Stevens R. C., GPCR Network
Science 2014, 344,58-64.
Further Optimization of the mGlu1 PAM VU6024578/BI02982816: Discovery and Characterization of VU6033685
Reed C. W., Kalbfleisch J. F., Turkett J. A., Trombley T. A., Spearing P. K., Haymer D. H., Quitalig M., Dickerson J. W., Foster D. J., Blobaum A. L., Boutaud O., Cho H. P., Niswender C. M., Rook J. M., Priepke H., Sommer H., Scheuerer S., Ursu D., Conn P. J., Melancon B. J., Lindsley C. W.
ACS Chem Neurosci 2025, 16(4), 745–752.
When you plan a publication, please use the following acknowledgement:
BI02982816 was kindly provided by Boehringer Ingelheim via its open innovation platform opnMe, available at https://www.opnme.com.