S-Bepafant
S-Bepafant WEB2388
S-Bepafant, the eutomer of racemic Bepafan, is a specific synthetic antagonist of the pro-inflammatory platelet activating factor (PAF) receptor. A pharmacologically improved derivative of Apafant, is the most potent derivative of thienotriazolodiazepine scaffolds. Is used in vitro and in vivo, The negative control is the inactive distomer WEB2387.



More information
The platelet-activating-factor receptor (PAFR) is a G-protein-coupled seven-transmembrane receptor that plays a profound role in stimulating inflammatory and thrombotic responses. PAFR is activated by platelet-activating-factor (PAF), which comprises a family of structurally related agonistic phospholipids that bind with high affinity to the receptor. PAFR stimulation mediates numerous cellular responses such as activation of the mitogen-activated protein kinase (MAPK) pathway, phosphoinositol turnover, platelet and granulocyte aggregation, and chemotaxis of leukocytes. PAF levels are elevated in disease tissues and fluids that lead to, amongst others, systemic hypotension, increased vascular permeability and thrombocytopenia. The interest in PAFR as a therapeutic target by inhibiting its function is underlined by its association with over 40 disease states that range from asthma to cancer. A number of diverse antagonists and inverse agonists of PAFR have been described that are either based on the original phospholipid structures or natural products, or entirely novel synthetic scaffolds. S-Bepafant represents a potent and well-characterised member of the latter class3,6,7,8.

PAF receptor in complex with the ligand SR 27417, indicating the presumed binding location of Apafant and Bepafant, as determined by X-ray crystallography (PDB code 5ZKP, Nat Struct Mol Biol 25: 488-495, 2018)
S-Bepafant binds with high affinity to the PAF receptor on human platelets, as determined by displacement of the natural ligand PAF from the PAFR receptor complex. Moreover, PAF-induced aggregation of both human platelets is inhibited by S-Bepafant in a dose-dependent manner. Whilst not specifically tested, the advantageous properties observed for racemic Bepafant will likely also apply to S-Bepafant, such as a lack of activity towards the benzodiazepine receptors.
In competition experiments with [3H]PAF, S-Bepafant displaces the natural ligand PAF with an equilibrium dissociation constant (KD) of 14 nM, thereby inhibiting the signaling function of PAFR. PAF-induced human platelet aggregation is inhibited in vitro at an lC50 of 350 nM.
| APAFANT | BEPAFANT | S-BEPAFANT | NEGATIVE CONTROL WEB2387 |
| MW [Da, free base]a | 455.97 | 467.97 | 467.97 | 467.97 |
| Assay A: Receptor Binding (KD) [nM], humanb | 152 | 169 | 149 | 6609 |
| Assay B: Platelet aggregation (IC50) [nM], humanc | 1701,11 | 3109,11 | 3509 | 87909 |
| Assay C: Neutrophil aggregation (IC50) [nM], humand | 3601 | 83010 | n.d. | n.d. |
| Assay D: Benzodiazepine receptor inhibition (Ki) [nM], rate | 3882 | 34952 | n.d. | n.d. |
a For the salt form you will get, please refer to the label on the vial and for the molecular weight of the salt, please refer to the FAQs
b Tritiated [3H]PAF binding to human platelets was inhibited by addition of increasing concentrations of Apafant, from which the KD was determined. In a reverse experiment, [3H]Apafant was displaced by PAF and Apafant to the same degree. Refer to respective references for detailed methods.
c Platelet-rich plasma isolated from human venous blood was collected, and aggregation was induced by addition of PAF. The aggregation inhibitory effect of the antagonists was determined adding various concentrations to the reaction mixture one minute prior to the addition of PAF. Refer to respective references for detailed methods.
d Human leukocytes were isolated from human venous blood. Aggregation was induced by addition of PAF, and the aggregation inhibitory effect of the antagonists was determined adding various concentrations to the reaction three minutes prior to the addition of PAF. Refer to respective references for detailed methods.
e Selectivity to benzodiazepine receptors was tested through inhibition of [3H]flunitrazepam binding to rat cortex synaptosomal membranes as a function of PAF antagonist concentration. Refer to respective references for detailed methods.
| APAFANT | BEPAFANT | S-BEPAFANT | NEGATIVE CONTROL WEB2387 | |
| Solubility @ pH 2.0/6.8 [µg/mL] | 55 / >100 | 33 / >100 | 51 / >100 | 44 / 86 |
| logD @ pH2 / pH11 | 1.08 / 1.12 | 1.21 / 1.15 | 1.2 / 1.14 | 1.18 / 1.12 |
| clogP | 0.98 | 0.87 | 0.87 | 0.87 |
| Plasma Protein Binding (%) human/rat | degradation / 65 | 54 / 33 | 38 / 34 | n.d. / n.d. |
| Caco-2 permeability AB @ pH 7.4 [*10-6 cm/s] | 3.2 | 11.8 | 7.1 | 15.1 |
| Caco-2 efflux ratio | 14.5 | 6.4 | 4.9 | 6.8 |
| Microsomal stability (human/rat) [% QH] | 24.9/38.3 | <23/25.4 | <23/24.3 | <23/25.1 |
| MDCK permeability PappAB @ 1µM [10-6 cm/s] | 0.25 | 1.1 | 0.94 | 0.72 |
| MDCK efflux ratio | 7 | 20.9 | 25.5 | 43.1 |
| Hepatocyte stability (human/rat) [% QH] | 20/54 | 7/55 | <4/48 | 6/58 |
| CYP 3A4 (IC50) [µM] | >50 | n.d. | >50 | n.d. |
| CYP 2D6 (IC50) [µM] | >50 | n.d. | >50 | n.d. |
| CYP 2C8 (IC50) [µM] | >50 | n.d. | >50 | n.d. |
| CYP 2C9 (IC50) [µM] | >50 | n.d. | >50 | n.d. |
| CYP 2C19 (IC50) [µM] | >50 | n.d. | >50 | n.d. |
| CODE | APAFANT | BEPAFANT | S-BEPAFANT | NEGATIVE CONTROL WEB2387 |
| tmax [h] rat (p.o.) | 0.3a | 0.8b | n.d. | n.d. |
| Cmax [nM] rat (p.o.) | 449 a | 491 b | n.d. | n.d. |
| Clearance [ml/(min*kg)]] | n.d. | 76 c | 44 d | n.d. |
| Mean residence time after iv dose [h] rat | n.d. | 0.38 | 0.5 | n.d. |
| F [%] | n.d. | 37b | n.d. | n.d. |
| VSS [l/kg] | n.d. | 1.7c | 1.3d | n.d. |
| t1/2 [h], rat, p.o. | 3.1a | 5.4b | n.d. | n.d. |
a p.o. dose: 5.3 mg/kg
b p.o. dose: 5.0 mg/kg
c i.v. dose: 0.48 mg/kg
d i.v. dose: 1.0 mg/kg
Acute bronchoconstriction induced by intravenously administered PAF is widely used to characterise PAF antagonists in animal models, where the antagonist efficacy is quantified by determining the recovery of respiratory flow and mean arterial pressure (MAP, a measure of hypotension).
In vivo, investigations using several animal models of human disease showed S-Bepafant to potently reduce bronchoconstriction and hypotension, where the efficacy exceeded that of racemic Bepafant. S-Bepafant and Bepafant represent pharmacologically improved derivatives of the previously described Apafant, showing higher potency in in vivo models1-3,9-11,13. S-Bepafant displays an ED50 of 0.018 and 0.004 mg/kg in guinea pigs when administered orally and intravenously, respectively, and the ED50 for MAP is comparable. Thus, the in vivo potency is similar or slightly improved compared to racemic Bepafant12, and significantly enhanced compared to Apafant. By contrast, negative control WEB2387, the distomer of Bepafant, shows a 40-80-fold reduction of in vivo potency, supporting the argument for improved potency of S-Bepafant versus racemic Bepafant12.
S-Bepafant is offered as the most potent derivative of the thienotriazolodiazepine class. The PAFR antagonists, Apafant and Bepafant are also available on opnMe.
| PROBE NAME / NEGATIVE CONTROL | APAFANT | BEPAFANT | S-BEPAFANT | NEGATIVE CONTROL WEB2387 |
| Respiratory flow ED50 [mg/kg] p.o. | 0.07 | 0.021 | 0.018 | 1.55 |
| Respiratory flow ED50 [mg/kg] i.v. | 0.018 | 0.007 | 0.004 | 0.081 |
| Mean arterial pressure ED50 [mg/kg] p.o. | 0.066 | 0.02 | 0.027 | 1.2 |
| Mean arterial pressure ED50 [mg/kg] i.v. | 0.016 | 0.006 | 0.005 | 0.086 |
Various additional pharmacology studies on racemic Bepafant are reviewed in reference 2.
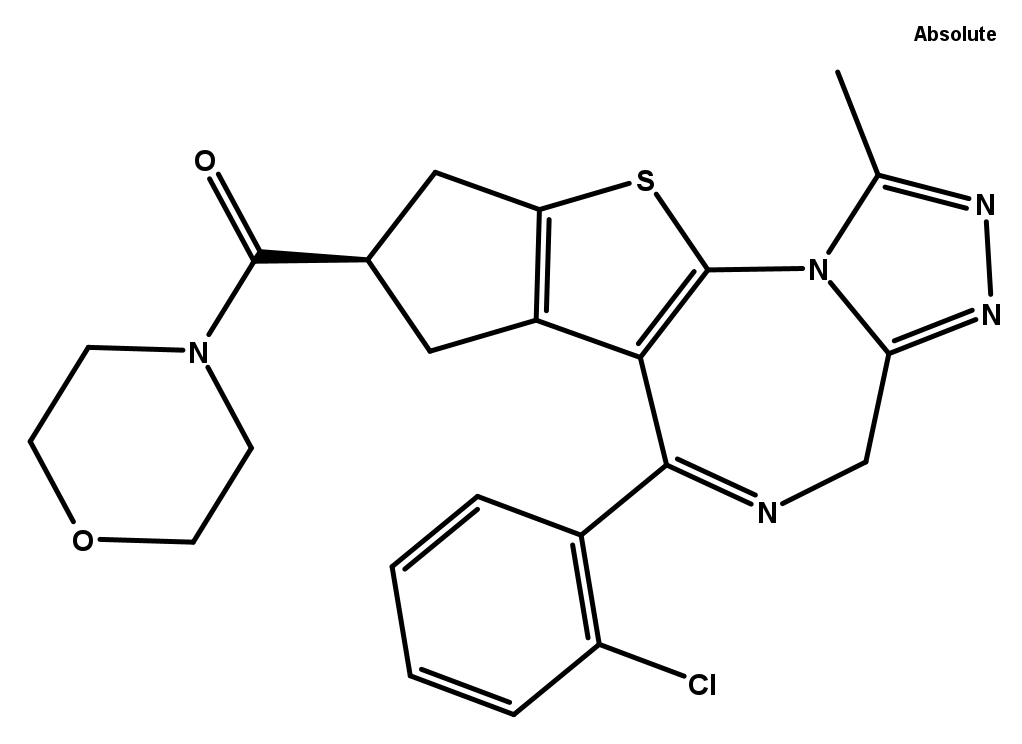
WEB2387 is offered as a negative control. It is the inactive R-isomer of Bepafant, and the mirror image of active S-Bepafant.
Structure of WEB2387, offered as an appropriate negative control
The SafetyScreen44™ panel has been measured only for Bepafant and it showed no relevant off-target effects.
| SELECTIVITY DATA AVILABLE | APAFANT | BEPAFANT | S-BEPAFANT | NEGATIVE CONTROL WEB2387 |
| SafetyScreen44™ with kind support of | Yes | Yes | Yes | Yes |
| Invitrogen® | No | No | No | No |
| DiscoverX® | No | No | No | No |
| Dundee | No | No | No | No |
Download selectivity data:
WEB2388_selectivityData.xlsx
WEB2387BS_selectivityData_1.xlsx
S- Bepafant is a synthetic platelet-activating-factor receptor (PAFR) antagonist based on the thieno-triazolodiazepine scaffold that has become a mainstay of in vitro and in vivo studies of the PAF pathway. S-Bepafant is offered as the most potent derivative of the thienotriazolodiazepine class. S-Bepafant represents the active isomer of Bepafant and displays somewhat enhanced in vivo potency compared to racemic Bepafant. As with racemic Bepafant, S-Bepafant binds with low nanomolar affinity to PAFR, and by competing with the natural ligand PAF, the proinflammatory function of the receptor is inhibited. We also provide the PAFR antagonists, Apafant and Bepafant. The inactive distomer WEB2387 is available as a negative control.
{kind=link}
{kind=link}
Pharmacological actions of WEB 2086, a new specific antagonist of platelet activating factor
Casals-Stenzel J, Muacevic G, Weber KH
J Pharmacol Exp Ther 1987, 241, (3), 974-81.
Pharmacodynamics, pharmacokinetics and safety profile of the new platelet-activating factor antagonist apafant
Brecht HM, Adamus WS, Heuer HO, Birke FW, Kempe ER
man. Arzneimittelforschung 1991, 41(1):51-9.
Platelet Activating Factor Antagonists
Summers JB, Davidsen SK, Sheppard GS
Current Pharmaceutical Design 1995, 1, 161-190.
Biological characterization of the enantiomeric hetrazepines of the paf-antagonist web 2170
Heuer H, Birke F, Brandt K, Muacevi G, Weber KH
Prostaglandins 1988, (35)5, 847.
Pharmacologic activity of bepafant (WEB 2170), a new and selective hetrazepinoic antagonist of platelet activating factor
Heuer HO, Casals-Stenzel J, Muacevic G, Weber KH
J Pharmacol Exp Ther 1990, 255(3):962-8.
Hetrazepins and processes for their preparation (EP024245)
13) Bechtel W-D, Casals-Stenzel J, Harreus A, Heuer H, Muacevic G, Stransky W, Walther G, Weber KH
1988
When you plan a publication, please use the following acknowledgement:
PAF Receptor Antagonist was kindly provided by Boehringer Ingelheim via its open innovation platform opnMe, available at https://www.opnme.com.